
Microbiome Technologies and Products


“微生物组(microbiome)是特定时间特定生境中微生物群所包含的基因序列(含同源序列)的总和。
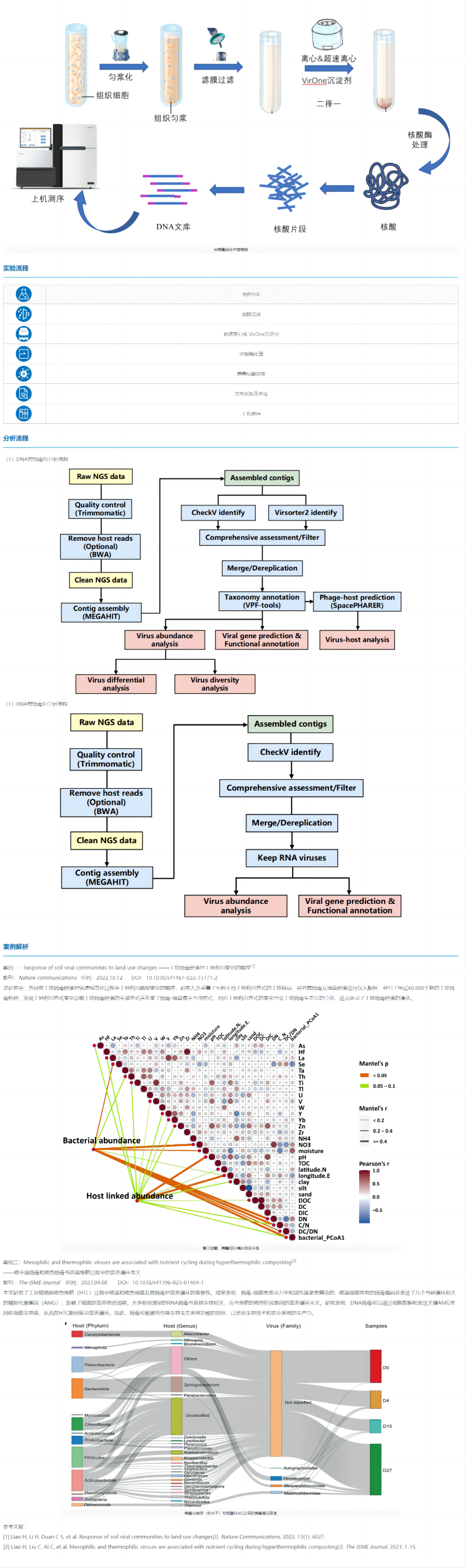
Viral metagenome, also known as Virome, is a new branch of discipline based on the theory of metagenomics and combined with existing virology and molecular biology techniques.
Macrovirome breaks through the limitations of traditional technical methods, directly takes the genetic material of all viruses in the environment or organization as the research object, and can quickly and accurately identify all virus components in the sample, which is a powerful means to discover new viruses, study the structure and diversity of viral populations, monitor virus mutation and evolutionary research. It plays an important role in virus discovery, virus traceability, and microbial early warning.
Future Research Trends
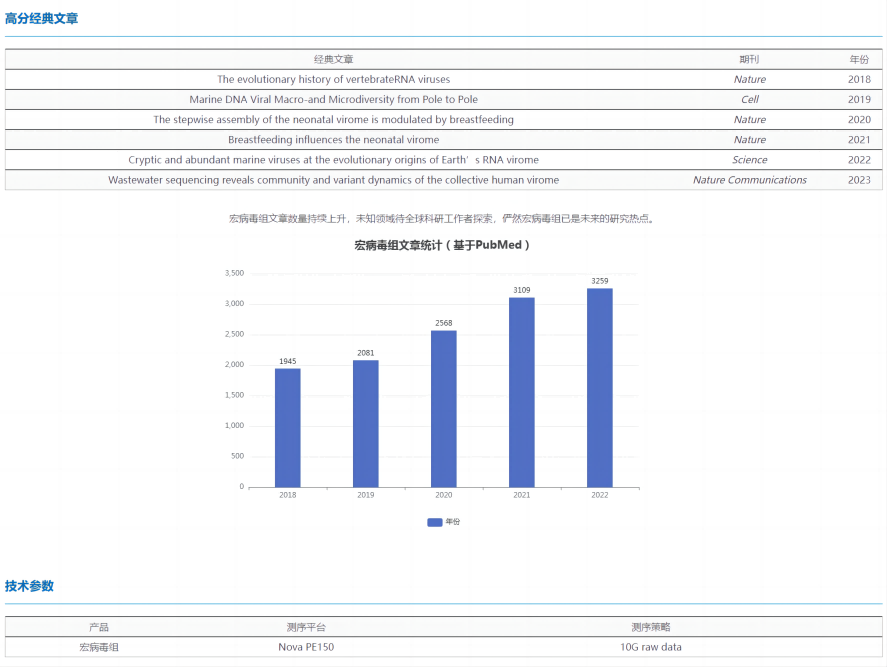
In recent years, many high-scoring articles on the macrovirome have been published, including soil, water and human body, which shows that the macrovirome has attracted the attention of researchers from all walks of life, and as the goal of follow-up research, the so-called "small viruses are everywhere; The virus has a great future!"
Technical route
Viruses are ubiquitous, but in human tissues, oceans, or soil samples, viruses are low in content due to their small genomes, and host components are difficult to remove in complex samples, so viral enrichment is a problem for the macrovirome. The scientific R&D team of Zhongtian Vaineng has developed two efficient virus enrichment solutions to help researchers overcome difficulties and accelerate the research process of macrovirome!
Ultracentrifugation: With the help of an ultracentrifuge, the sucrose density gradient centrifugation of the preliminarily filtered sample for more than 24 hours can effectively separate the virus particles in the sample from prokaryotes and cell debris.
VirOne Competitive Precipitation: The original VirOne competitive precipitation method competitively binds water molecules and neutralizes surface charges, forcing VLPs to precipitate from solution and rapidly collecting viral particle pellets by conventional centrifugation.
Both of the above schemes have been tested by a large number of samples, which can greatly increase the proportion of effective virus data in the sequencing data, and provide a reliable guarantee for the subsequent analysis results.
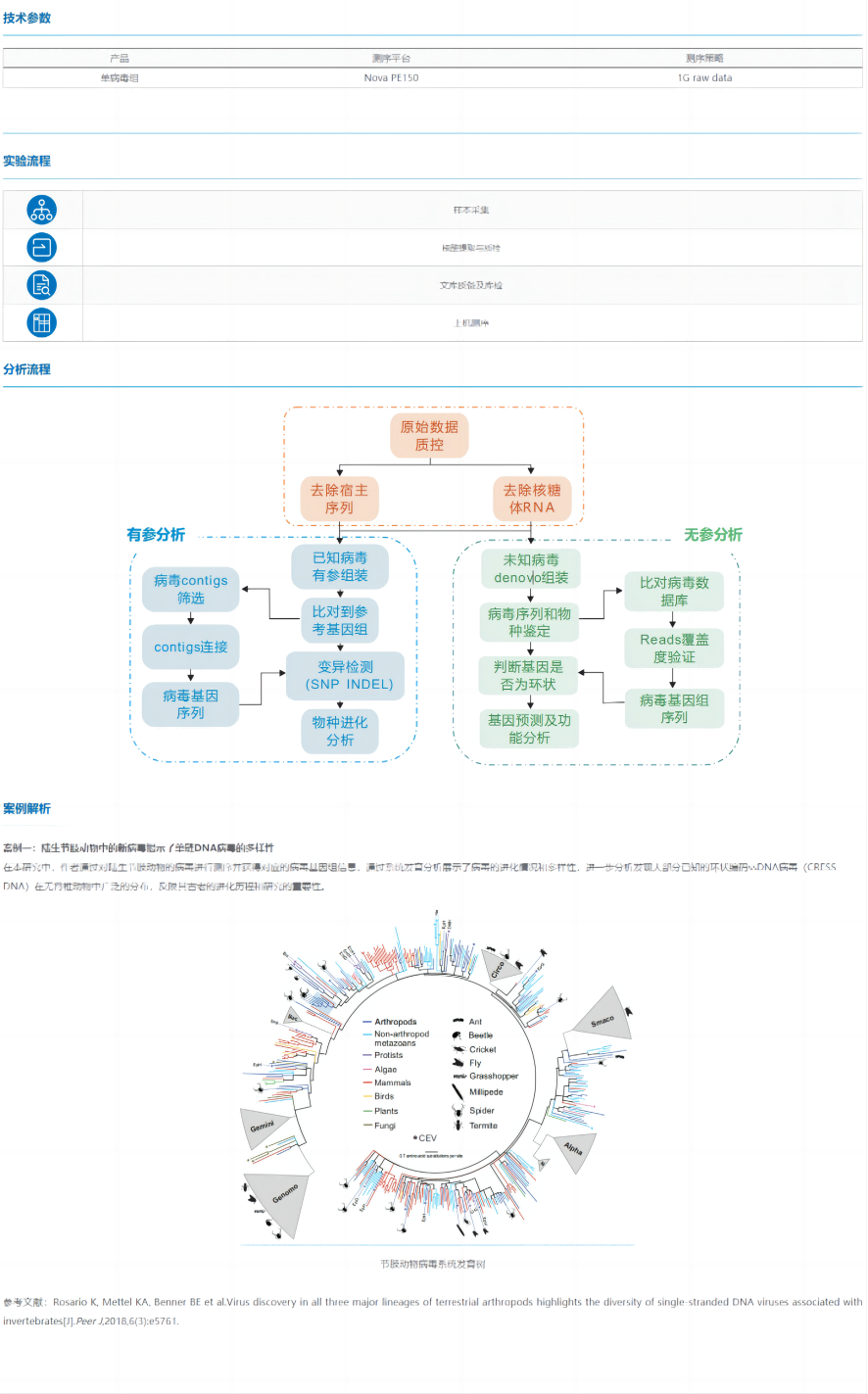
The single viral genome takes the virus in culture or natural samples as the research object, first isolates a single viral particle and performs whole genome amplification, obtains genome sequence information to identify the virus, and configures the corresponding analysis process according to the reference genome of the target virus.
Whole-genome sequencing analysis of viruses will greatly facilitate the identification of emerging infectious diseases, the development of rapid detection reagents, the development of vaccines and drugs, and the estimation of transmission routes. In addition, it is important to improve our understanding of viral diversity, adaptability, ecological properties, and evolution.
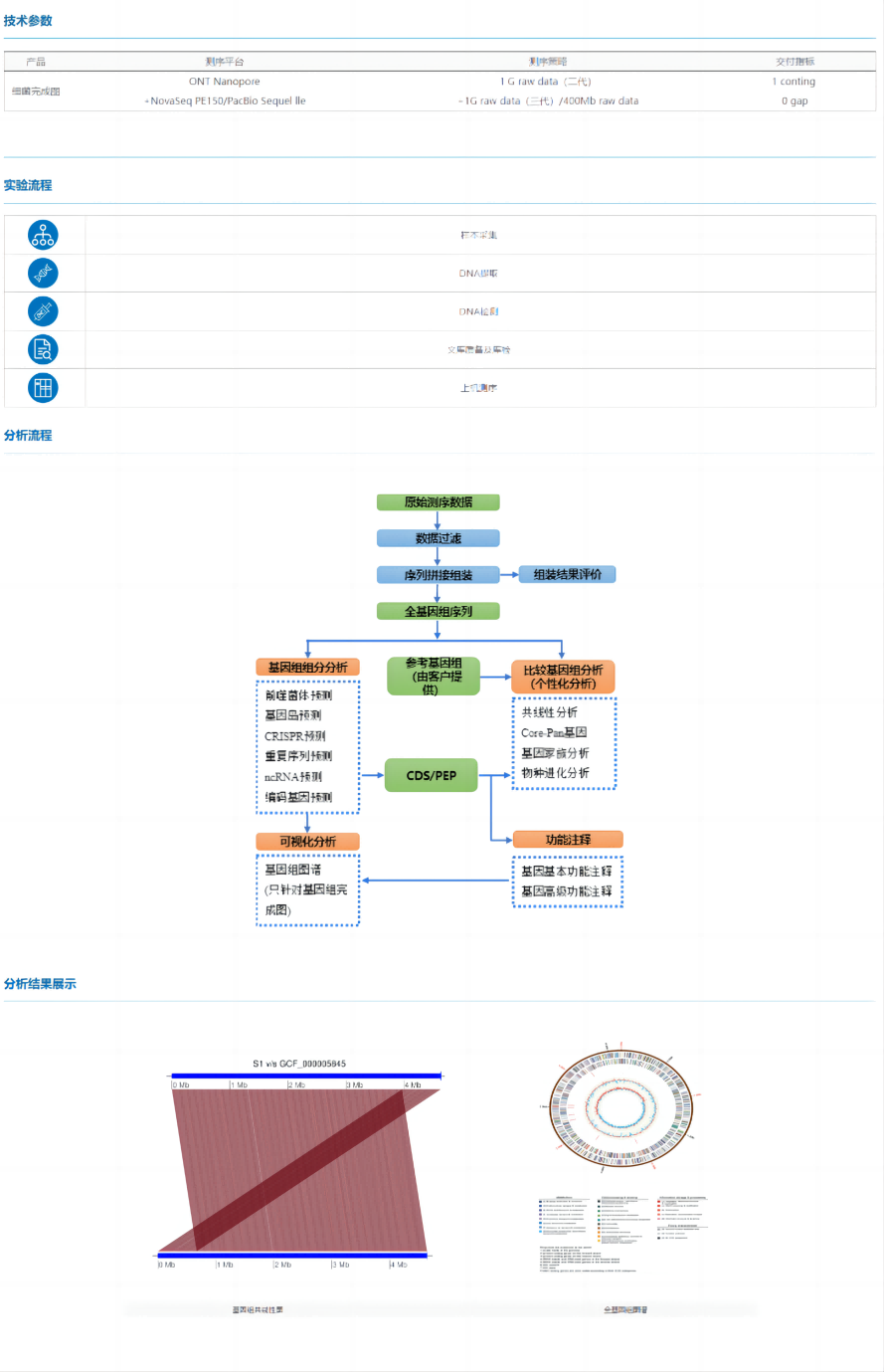
Bacterial completion map refers to the comprehensive use of second-generation or third-generation sequencing technology to carry out genome sequencing and assembly according to the specific information of the strain, and finally obtain a complete genome sequence without gap. The bacterial completion map can obtain comprehensive and rich genome information of bacteria, so as to dig deep into functional genes from multiple perspectives such as pathogenicity, drug resistance, and environmental adaptability, and conduct in-depth research on the characteristics of different phenotypes of microorganisms.
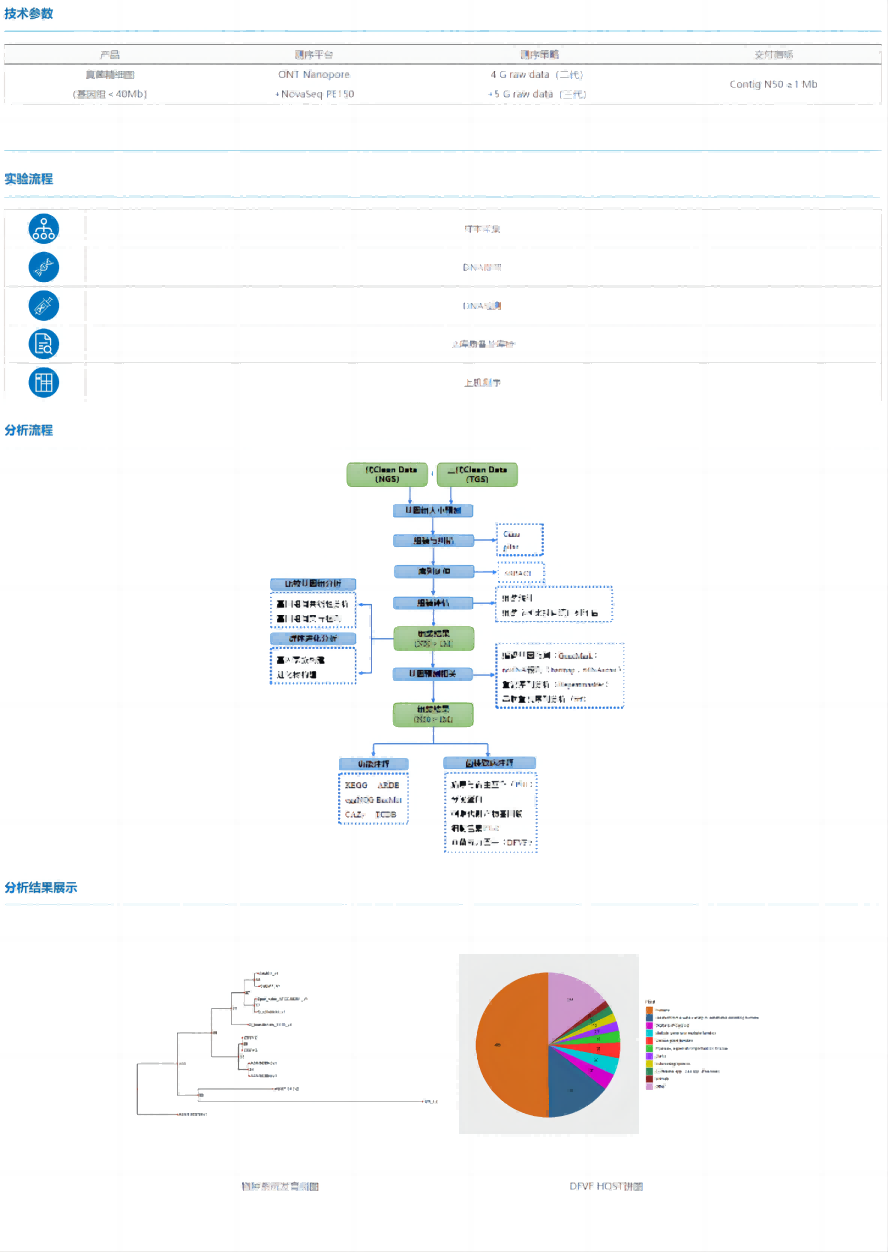
Fungal fine map refers to the use of second- and third-generation sequencing technology to obtain high-quality fungal genome assembly results, so as to achieve a more in-depth and systematic study of fungal genome structure and function.
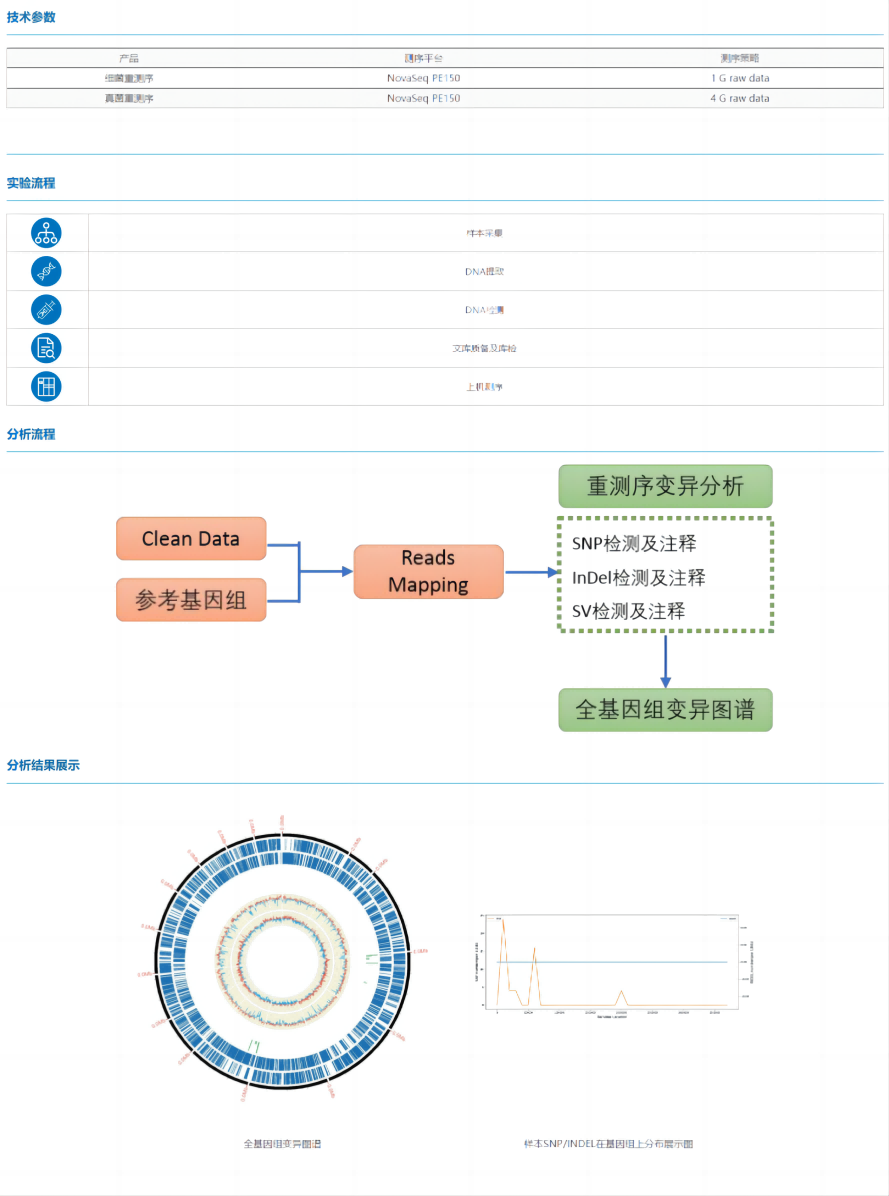
Microbial resequencing refers to a sequencing analysis technology that performs high-throughput data analysis on a microorganism by comparing it with a close reference genome. Through resequencing, a series of variation information such as SNP, InDel, and SV of the target microbial genome from the reference genome can be obtained, and the trait differences between the genomes can be predicted and analyzed, so as to complete the study of the target microorganisms.
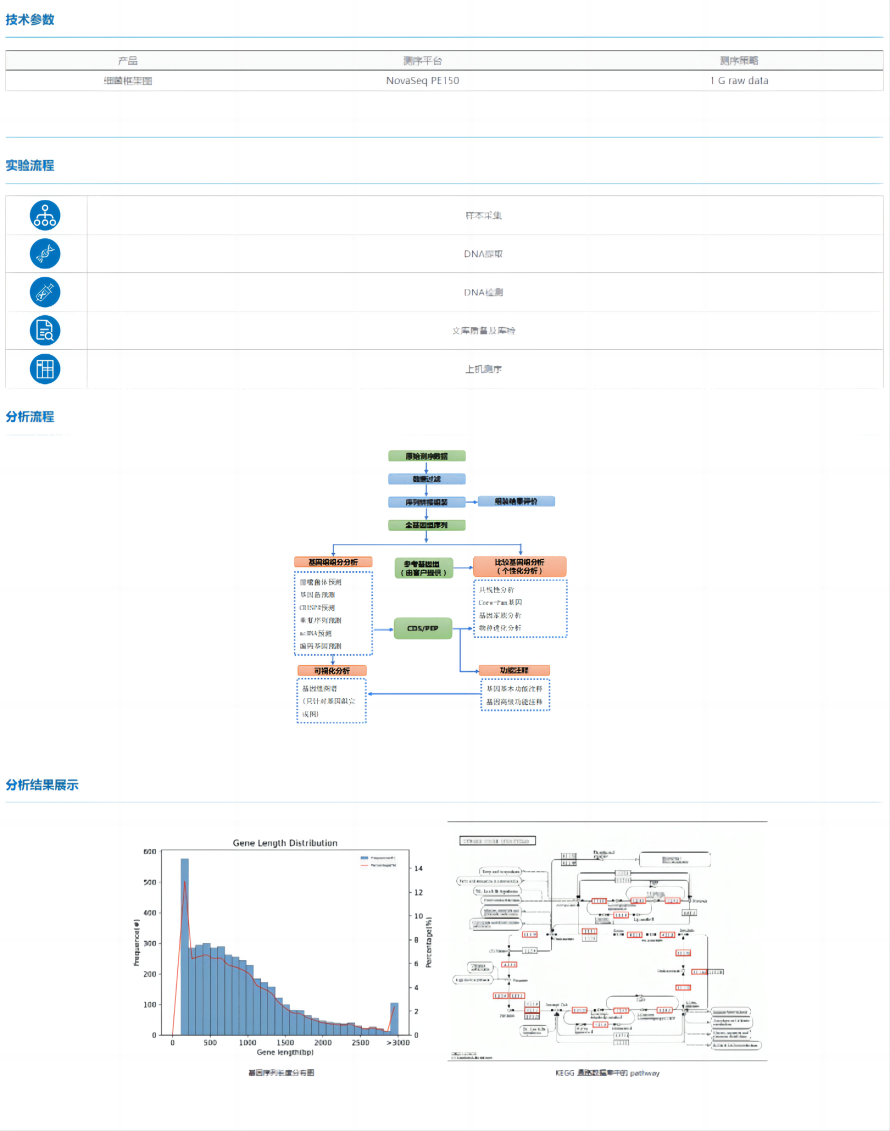
Bacterial framework diagram is a relatively preliminary method of bacterial whole-genome sequencing, in which the bacterial genome is sequenced and the corresponding assembly steps are performed by the second-generation Illumina sequencer, and finally the bacterial genome sketch is obtained. The bacterial framework map project is cost-effective and can meet the basic needs of bacterial genome research.
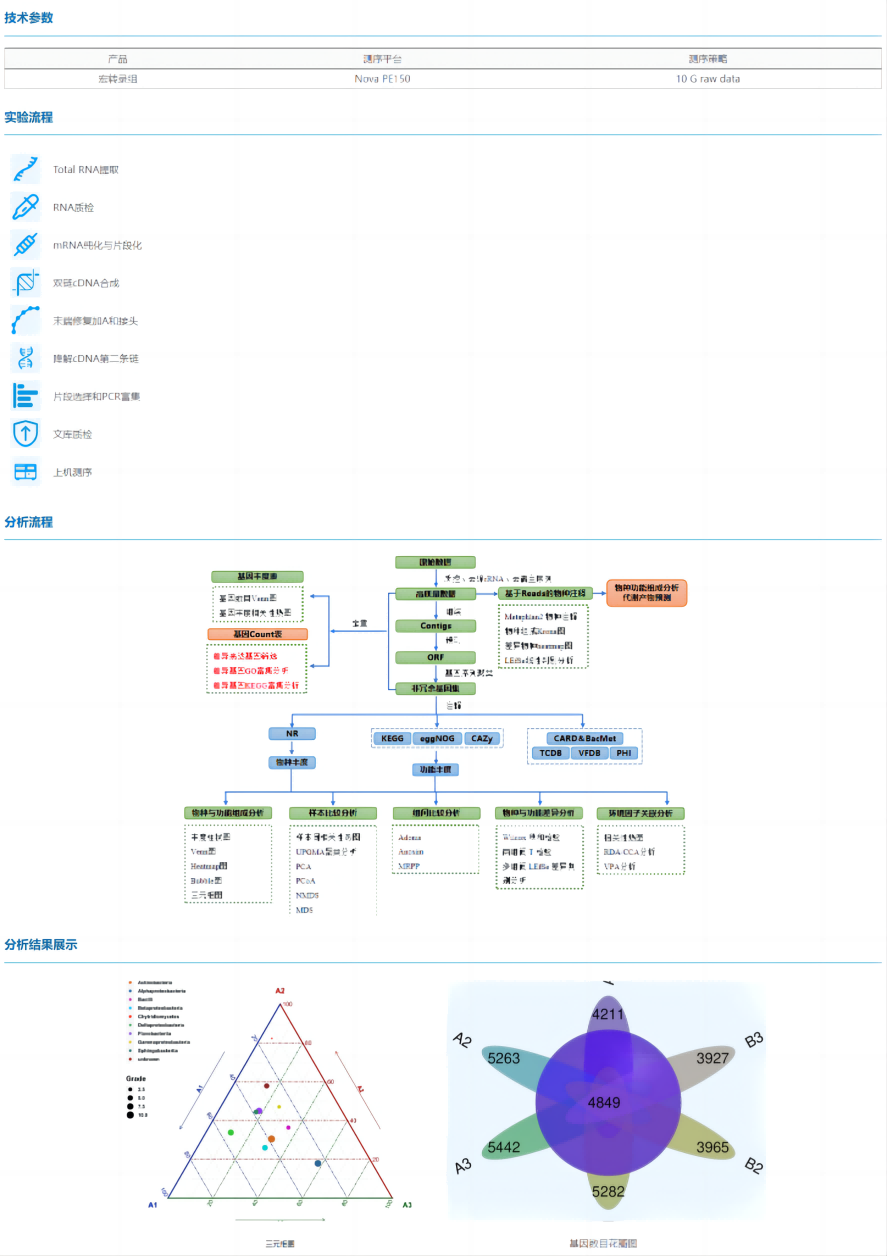
Metatranscriptome sequencing is the study of the entire microbial community in a specific environment, which gets rid of the technical limitations of microorganisms that are difficult to isolate and culture, and only needs to extract the total RNA of all microorganisms from the sample for sequencing.
Metatranscriptome sequencing uses the Novaseq second-generation high-throughput sequencing platform to comprehensively sequence the RNA of microbial communities in the samples, and through assembly, gene prediction, gene annotation and abundance calculation, the genetic diversity and molecular ecological information such as species composition, transcriptional regulation of functional genes and metabolic pathways in the samples are mined.
Features:
l Microbial communities can be identified at the transcriptional level and community characteristics can be studied;
l Identification of target pathogens;
l Excavation of live microorganisms and their functions;
l Focus on key genes and explore gene function and metabolic pathways.
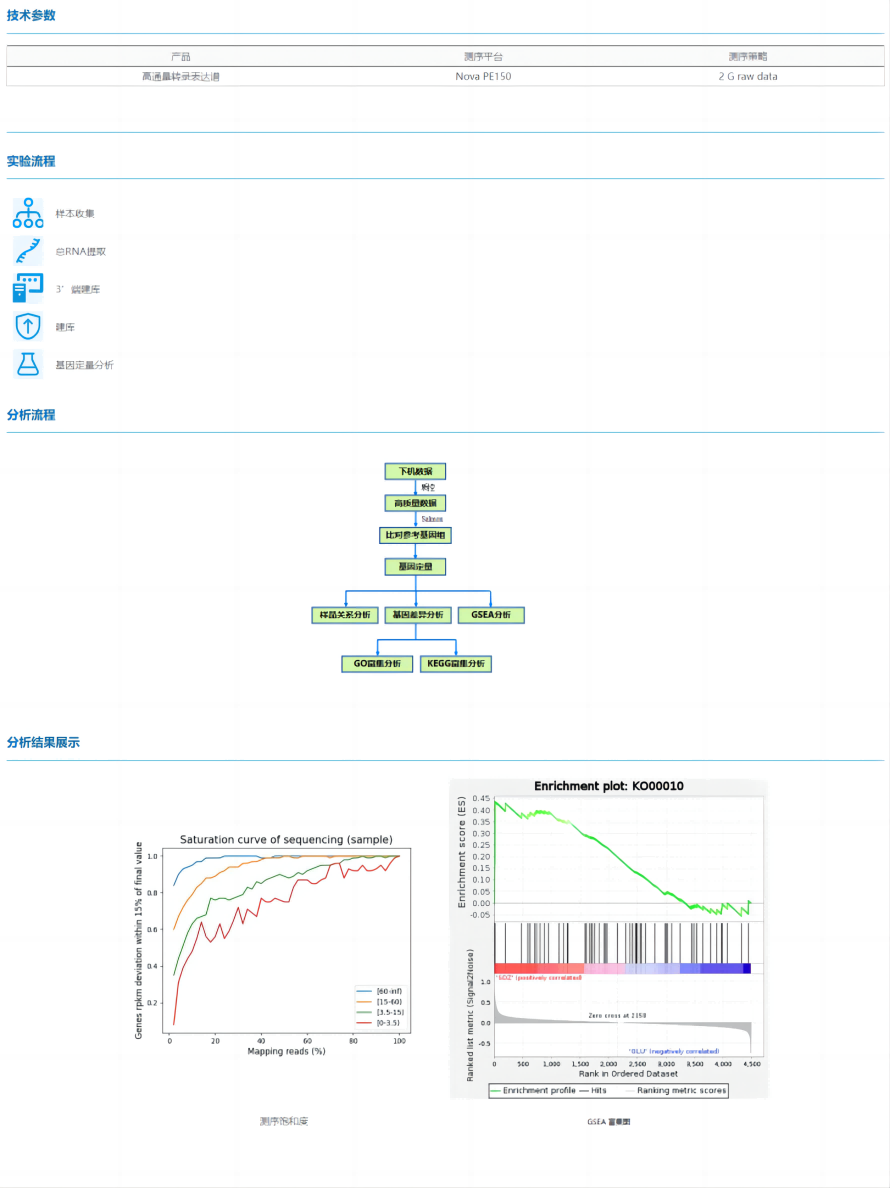
High-throughput transcription and expression profiling (HT-RNAseq) is a eukaryotic transcriptome product based on the mature 3' end library construction technology in the single-cell transcriptome, which can simplify the library preparation experiment and save the library construction cost, sequence the sequence near the 3' end of mRNA and quickly obtain gene quantification and differential analysis results, which is suitable for most eukaryotic samples, even FFPE samples.
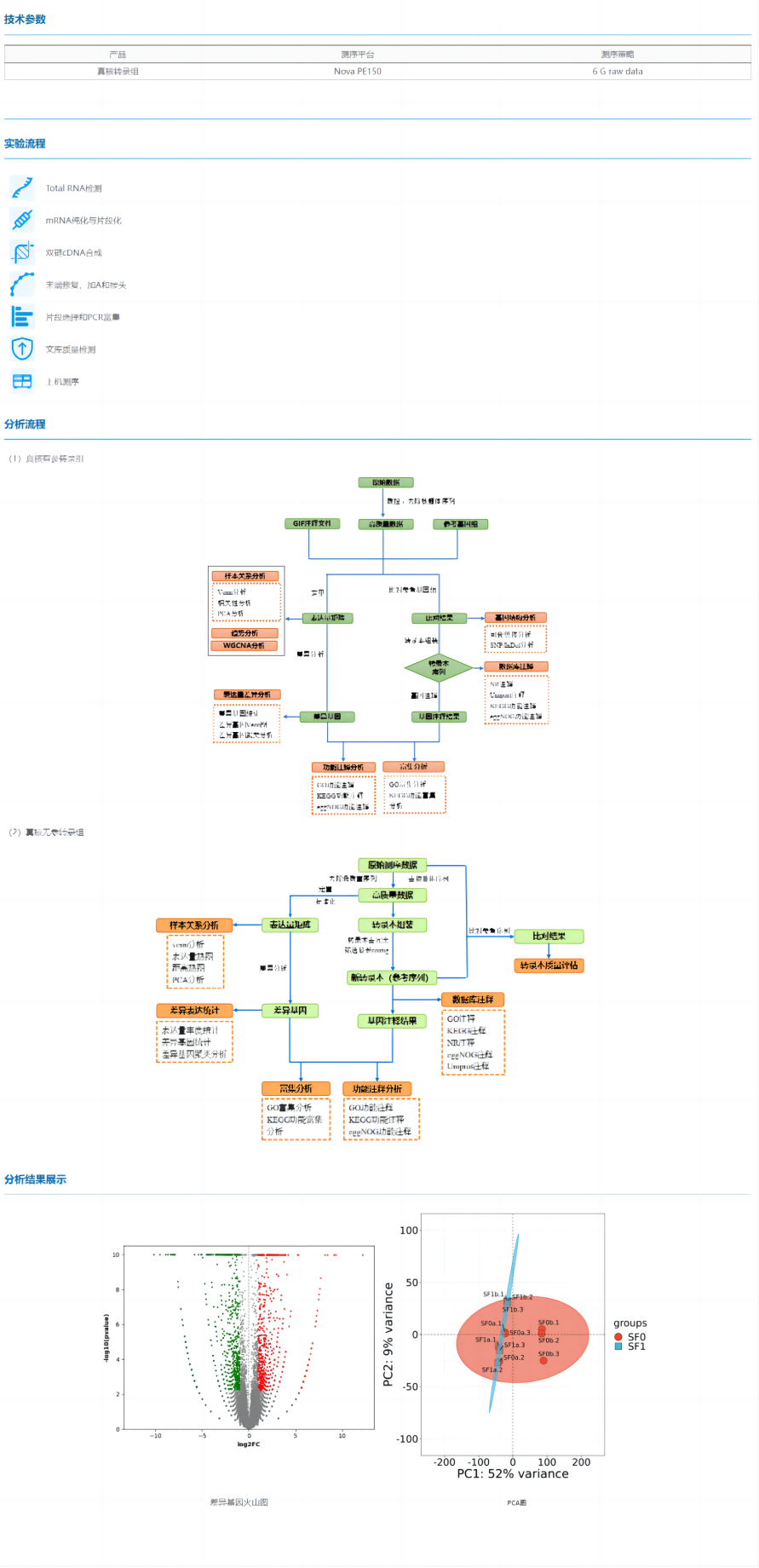
The eukaryotic transcriptome is mainly divided into two categories according to whether there is a reference genome or not, the eukaryotic reference transcriptome and the eukaryotic non-reference transcriptome.
Eukaryotic reference transcriptome: refers to the transcriptome sequencing of eukaryotes or their tissues and cells that have a reference genome. The sequencing results can be compared with the reference genome to verify the sequencing saturation and gene coverage of transcriptome sequencing, and obtain information such as gene structure, gene expression differences, and alternative splicing.
Eukaryotic parameter-free transcriptome: refers to the sequencing of eukaryotic transcriptome in the absence of a reference genome. In eukaryotic non-parametric transcriptome sequencing, firstly, after obtaining the original data, the data needs to be quality controlled and spliced into unigene, and then unigene is used as the reference sequence for subsequent analysis. Differential gene expression analysis and differential gene function enrichment analysis can also be performed on multiple samples to discover functional genes and provide directions for the next step of research.
Features:
l High coverage: comprehensive coding RNA information can be obtained in a single transcriptome sequencing experiment;
l Rich annotation information: through comparison with major databases, relatively complete transcriptome annotation information can be obtained;
l Correlation analysis: Multiple omics data can be combined to carry out cross-omics association analysis.
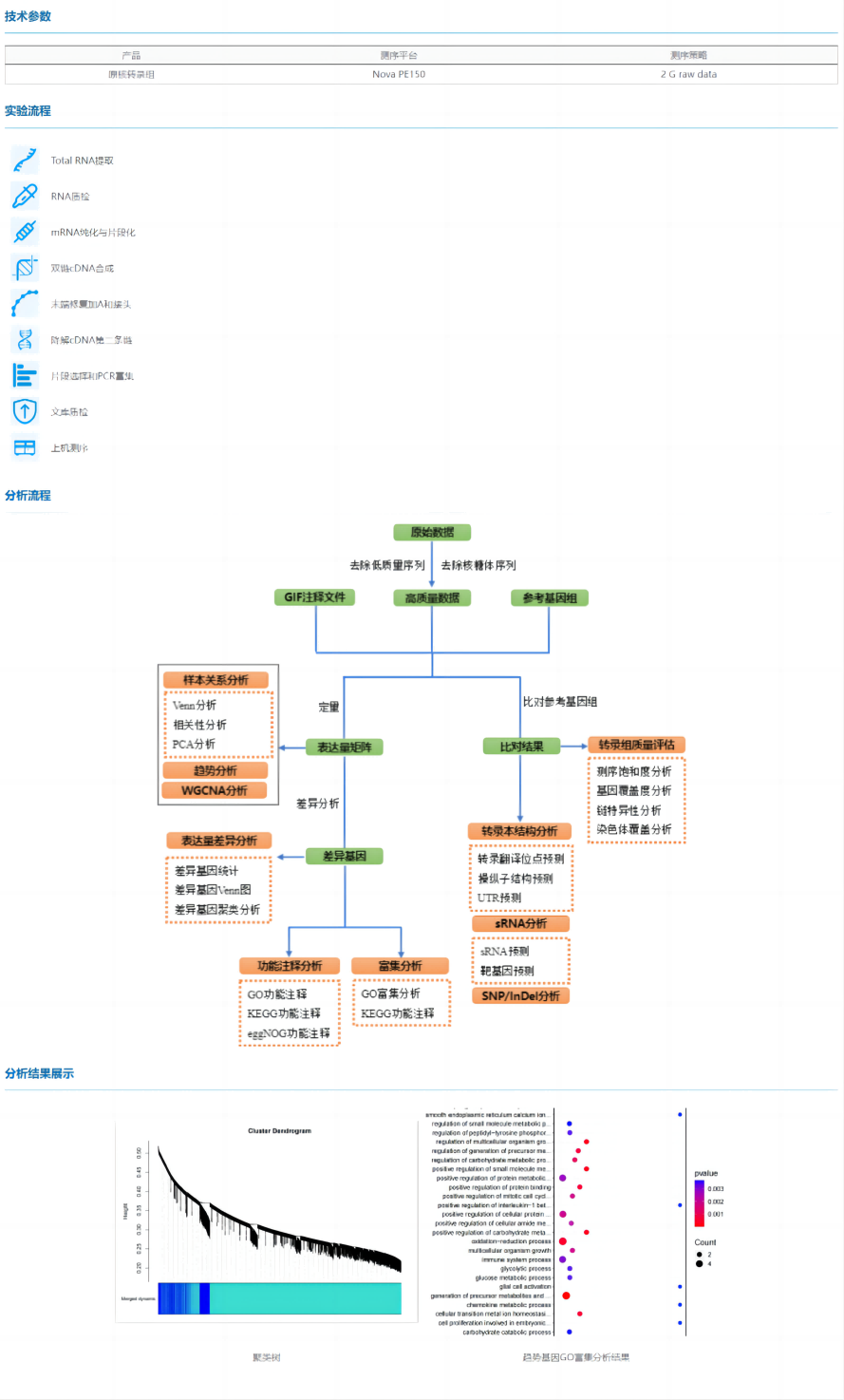
Prokaryotic transcriptome sequencing refers to the use of high-throughput sequencing technology to sequence the transcripts of prokaryotes, and comprehensively and quickly obtain the information of all transcripts of specific microorganisms in a specific state. Transcriptome sequencing can not only quantitatively analyze the expression of mRNA, analyze differentially expressed genes and their corresponding functions, but also analyze sRNAs to reveal the molecular regulatory mechanism and function of different phenotypes of microorganisms.
Features:
l The kit was used to remove rRNA, which had good rRNA removal efficiency and high data quality;
l Chain-specific library construction is adopted, and the stability of library construction is good;
l In addition to mRNA quantitative analysis, transcript structure analysis can also be performed;
l mRNA, sRNA prediction and variant analysis can be performed at the same time to more comprehensively interpret the expression interaction network.

|
24-hour service hotline:028-87712800 |

|
service email:sctchj@188.com |

|
company address:No. 2507, Building 1, No. 8, Yuren West Road, Jinniu District, Chengdu, Sichuan Province |