
Microbiome Technologies and Products


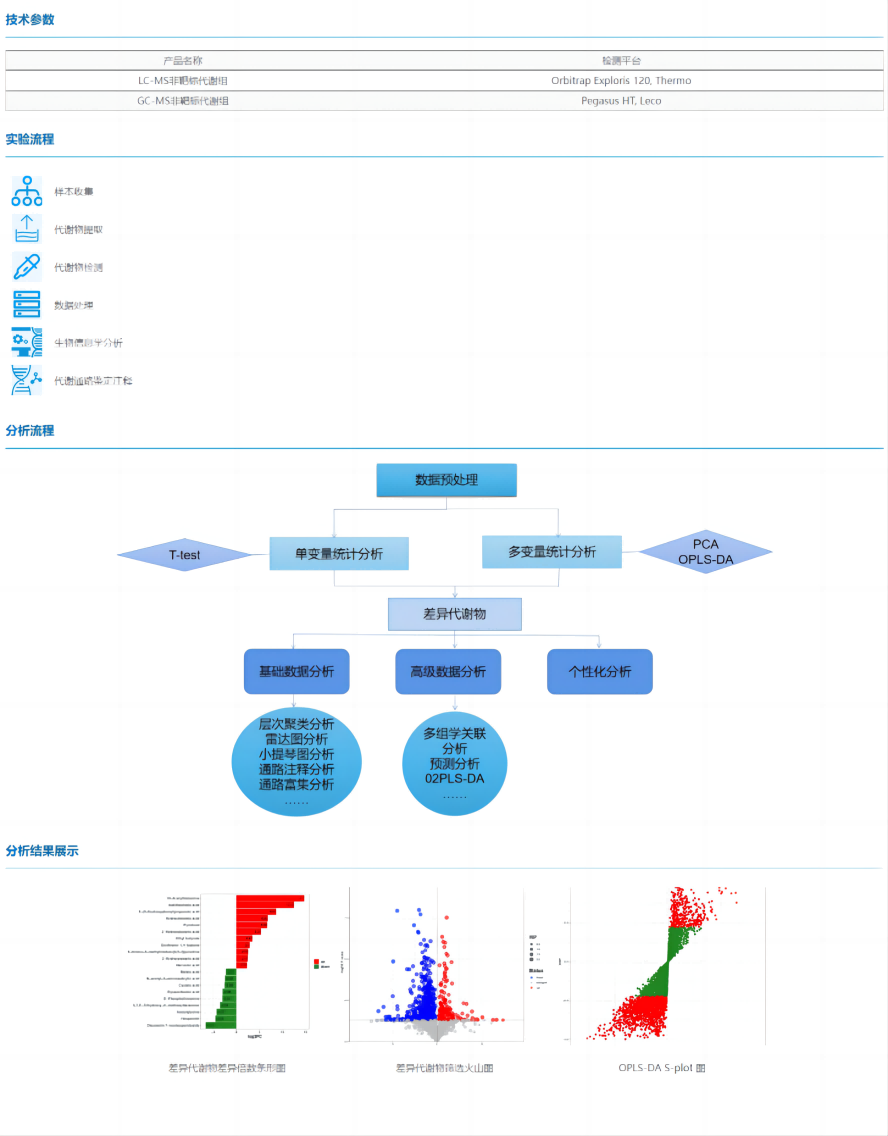
Non-target metabolomics is an unbiased metabolomic analysis that enables the discovery of new biomarkers or the discovery of differential metabolites for a comprehensive and systematic analysis of endogenous metabolites in an organism. Non-target metabolomics can detect a large number of metabolite signals simultaneously, but its sensitivity and qualitative quantification accuracy are poor. According to the different detection platforms, it can be subdivided into LC-MS non-target metabolomics and GC-MS non-target metabolomics.
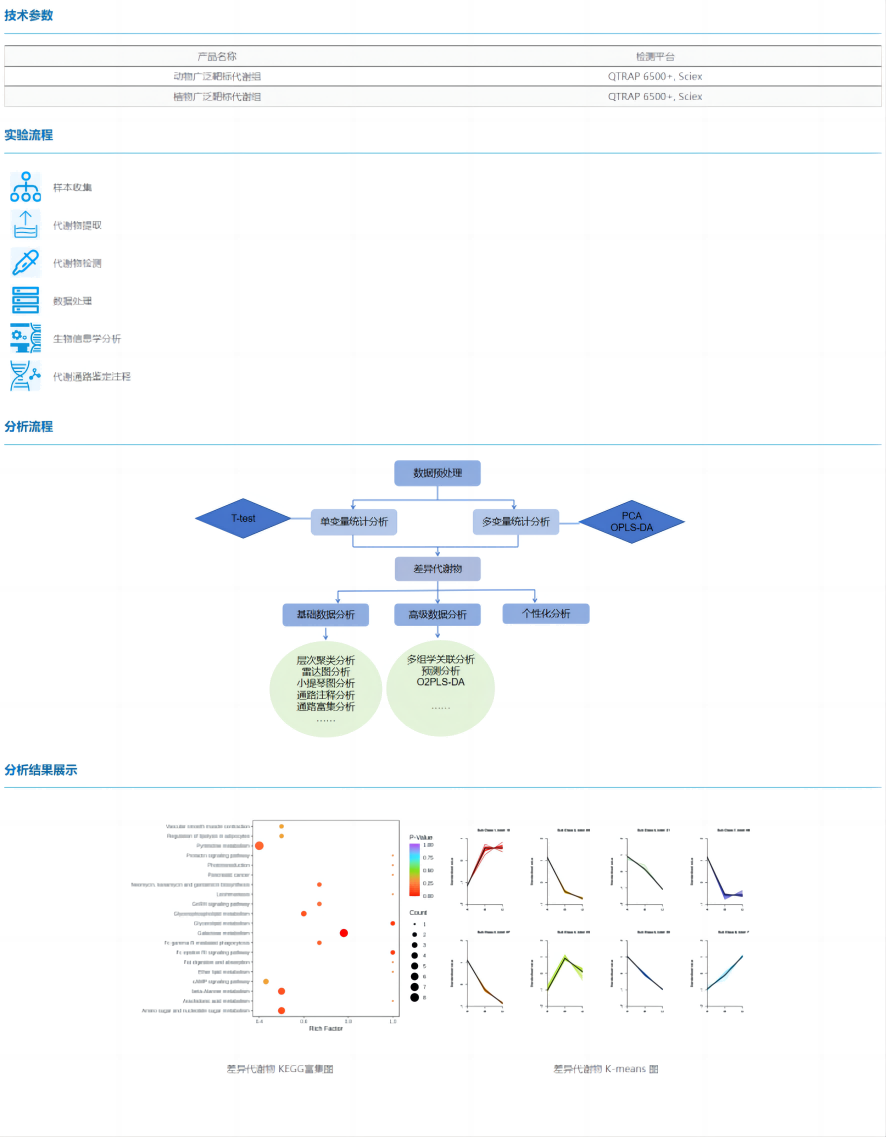
Broad-target metabolomics is a new type of metabolomics detection technology that integrates the advantages of "extensiveness" of non-target metabolome and "accuracy" detection technology of target metabolome, which has the characteristics of high throughput, ultra-sensitivity, wide coverage, qualitative and quantitative accuracy.

High-throughput target metabolomics integrates the technical advantages of non-target metabolomics and target metabolomics, creatively realizes high-throughput and high-sensitivity target metabolite detection, and provides an efficient method for qualitative and quantitative detection of large-scale and low-abundance metabolites.
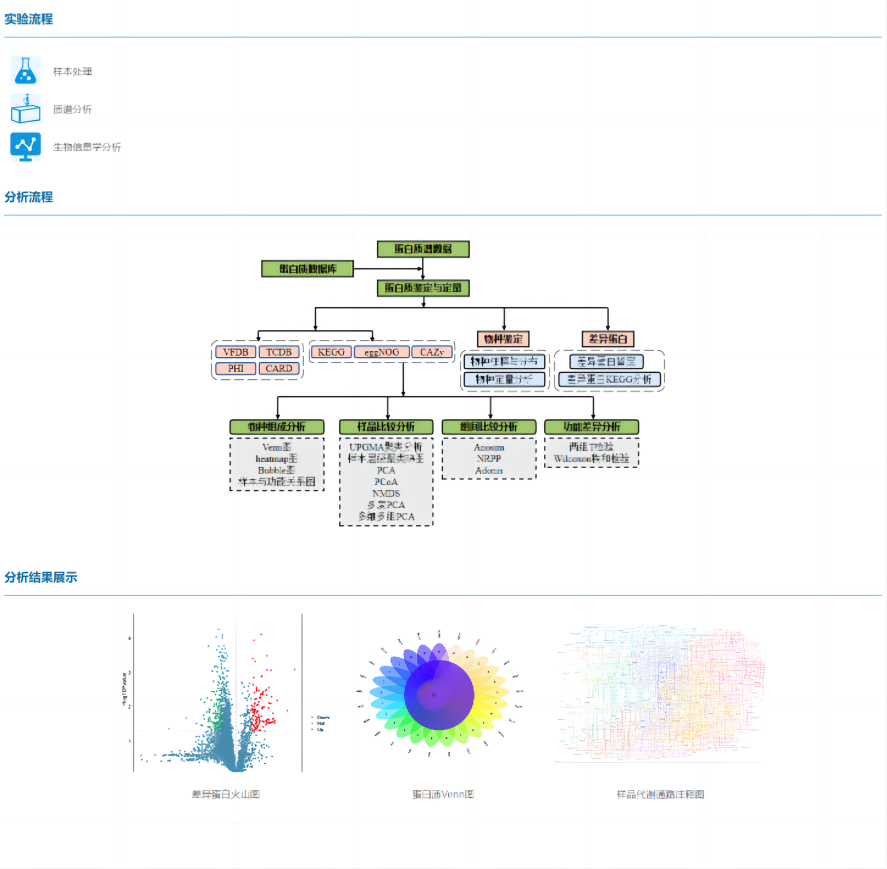
Metaproteomics is the large-scale qualitative and quantitative of proteins in microbial communities, thereby revealing microbial phenotypic information at the molecular level. It can use mass spectrometry technology to collect protein information of microbial populations in nature on a large scale, and combine a variety of omics data to carry out research on the genetic characteristics and biological functions of microbial populations.
Features:
l Targeted protein extraction method to improve the efficiency of macroprotein extraction;
l Dedicated protein database to improve the accuracy of protein identification;
l Advanced mass spectrometry analysis platform to produce high-quality data;
l Complete data analysis process, providing personalized data analysis.

General proteomics is a science that takes the proteome as the research object and studies the protein composition of cells, tissues or organisms and their changes. Proteomics essentially refers to the large-scale study of protein characteristics, including protein expression levels, post-translational modifications, protein-protein interactions, etc., so as to obtain a holistic and comprehensive understanding of the processes of disease occurrence and cellular metabolism at the protein level.
Research Methods:
(1)Label free
Protein label-free quantification technology (Label free) is a mass spectrometry analysis of protein digested peptides by liquid chromatography-mass spectrometry technology, without the need to use expensive stable isotope tags as internal standards, only need to analyze the mass spectrometry data generated when large-scale identification of proteins, and compare the signal intensity of the corresponding peptides in different samples, so as to relatively quantify the corresponding proteins of peptides.
Features:
l No need for isotope labeling, simple operation and low cost;
l Low identification coverage and sensitivity;
l Not limited by the number of samples;
l Protein "presence/absence" comparative analysis can be performed;
l High requirements for mass spectrometry instruments and equipment, chromatography and mass spectrometry need to have good stability and repeatability at the same time.
(2)DIA
DIA technology mainly uses the data-independent acquisition mode (DIA), combined with the traditional data-dependent acquisition mode (DDA) to construct a reference spectrum library, and divides the entire mass spectrometry scanning mass range into several windows through liquid chromatography-mass spectrometry (LC-MS/MS), and fragments all ions in each window in turn. Collect all product ion information. The obtained data were used in mainstream analysis software for proteomics identification, quantification and differential protein analysis. This technique eliminates the need to specify the peptide of interest, and can obtain all fragmentation information of all ions in the sample without missing any gaps, greatly improving data utilization.
Features:
l High identification sensitivity: more low-abundance proteins can be identified, improving the confidence of quantitative analysis;
l Improved repeatability: Compared with the DDA mode, there are fewer missing values of DIA and higher data reproducibility;
l Suitable for large sample size: it is not limited by sample size and can be used for large-scale sample size detection;
l High quantitative accuracy: the CV value is low, and the quantitative accuracy is improved;
l Data can be traced: Digitized protein information can be traced.
(3)Direct DIA
Compared with the traditional DIA analysis strategy, Direct DIA no longer performs DDA hierarchical database building, but uses machine deep learning to directly search for DIA original file spectra to generate a library. Deep learning scores to find peak fragmentation, predict retention time, and remove false positive results. Compared with DDA, it has the advantages of fewer missing values, a larger number of identified proteins, and accurate quantification. This significantly reduces the cost compared to traditional DIA techniques, and the number of proteins identified is getting smaller and smaller compared to traditional DIA.
Features:
l DIA holographic scanning, good data integrity, high protein coverage;
l Do not carry out DDA library construction, improve efficiency and reduce experimental costs;
l Few missing data values, good reproducibility effect, and accurate quantification.
(4)TMT
TMT technology is a quantitative proteomics method that enables the simultaneous quantification of proteins in multiple samples based on the principle of chemical labeling. This technique utilizes specific chemical reagents (TMT tags) to label proteins in different samples with different mass-to-charge ratios, which are then quantified by tandem mass spectrometry of a mass spectrometer.
Features:
l High throughput: TMT technology can analyze multiple samples at the same time to improve the analysis efficiency;
l High sensitivity: TMT technology has high sensitivity and can detect low-abundance proteins;
l High quantitative accuracy: TMT technology provides accurate quantitative results, which can be used for quantitative comparison and trend analysis.

|
24-hour service hotline:028-87712800 |

|
service email:sctchj@188.com |

|
company address:No. 2507, Building 1, No. 8, Yuren West Road, Jinniu District, Chengdu, Sichuan Province |