
Microbiome Technologies and Products


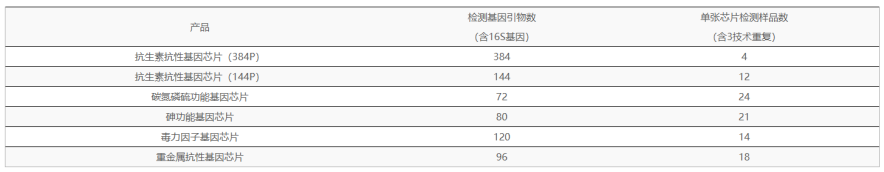
The 16S rDNA gene is a commonly used molecular marker in the phylogenetic classification of prokaryotic microorganisms, and is widely used in the study of microbial ecology. The quantitative method of high-throughput qPCR (HT-qPCR) microarray detection technology has overcome the barriers of sequencing omics methods in terms of co-efficacy and precise quantification, and has been widely used to describe and interpret the functional structure of microorganisms in various ecosystems.
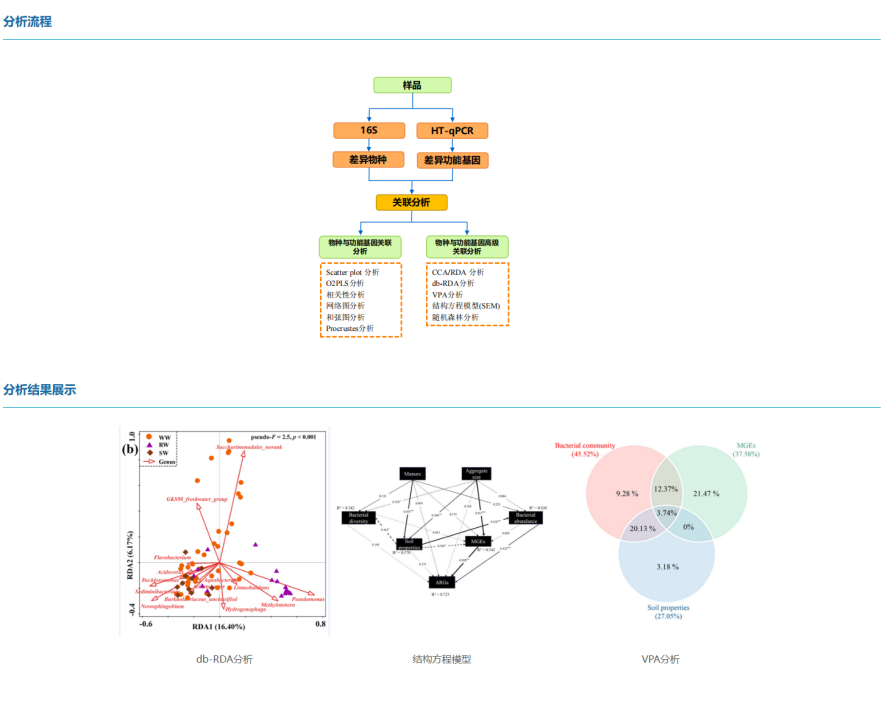
Based on the omics data of 16S and high-throughput qPCR chips, the microbial communities and functional genes with differences between groups were screened out from the level of 16S and functional genes, and then the correlation between the two abundances was jointly analyzed to reveal the correlation between specific microflora and specific functional genes from multiple perspectives, which is of great significance for the study of community structure and function.
Joint analytics benefits
l Mutual verification through omics analysis at the level of microflora and functional genes;
l At the same time, biological problems can be explored from the aspects of "cause" and "effect";
l Explain the association mechanism between microbiota and functional genes, and more comprehensively analyze the species and functional regulation mechanisms in biological processes.
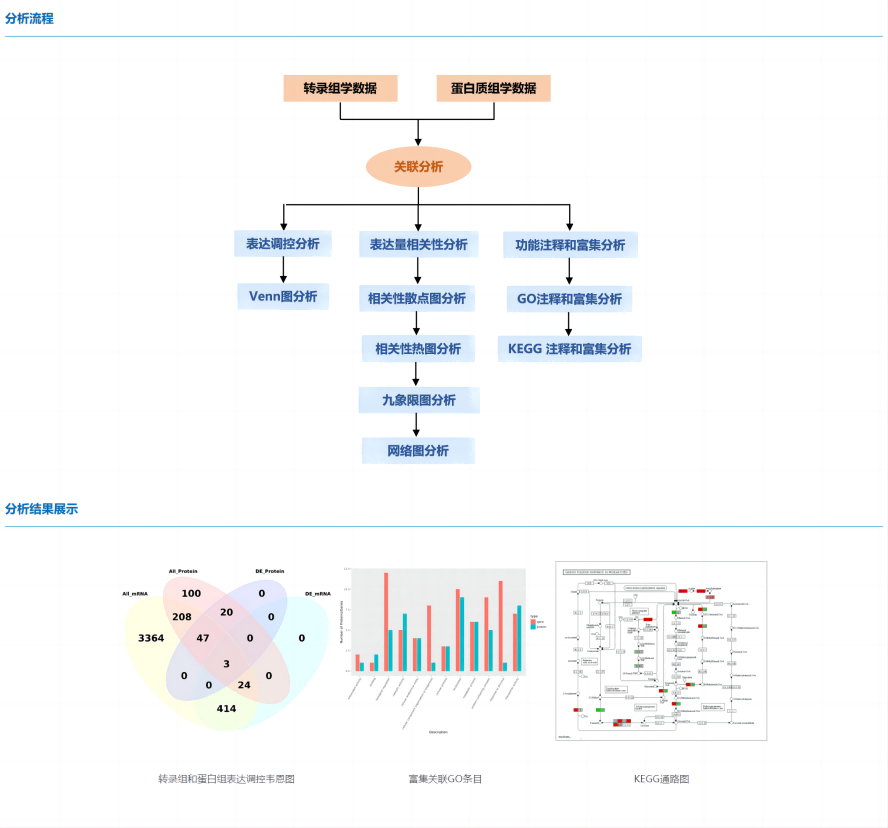
From a biological point of view, the mRNA level represents the intermediate state of gene expression, and the potential expression of protein can be speculated, and protein is the direct executor of life function, which has a direct and important role in the measurement of gene expression level. In this way, we can interpret the law and essence of life activities at different omics levels, explain biological problems as a whole, comprehensively explore the disease mechanism and stress mechanism of organisms, and accurately study the expression patterns and regulatory mechanisms of important genes.
Joint analytics benefits
l Transcriptome and proteomics data complement each other, allowing for all-round analysis of mRNA and protein expression levels in organisms in specific states;
l Through in-depth analysis of data, we can dig out differential genes or differential proteins, and find and verify some important regulatory pathways;
l For those species with lack of protein database or incomplete annotation, constructing a protein search library from transcriptome data can greatly improve the number and quality of protein identification.
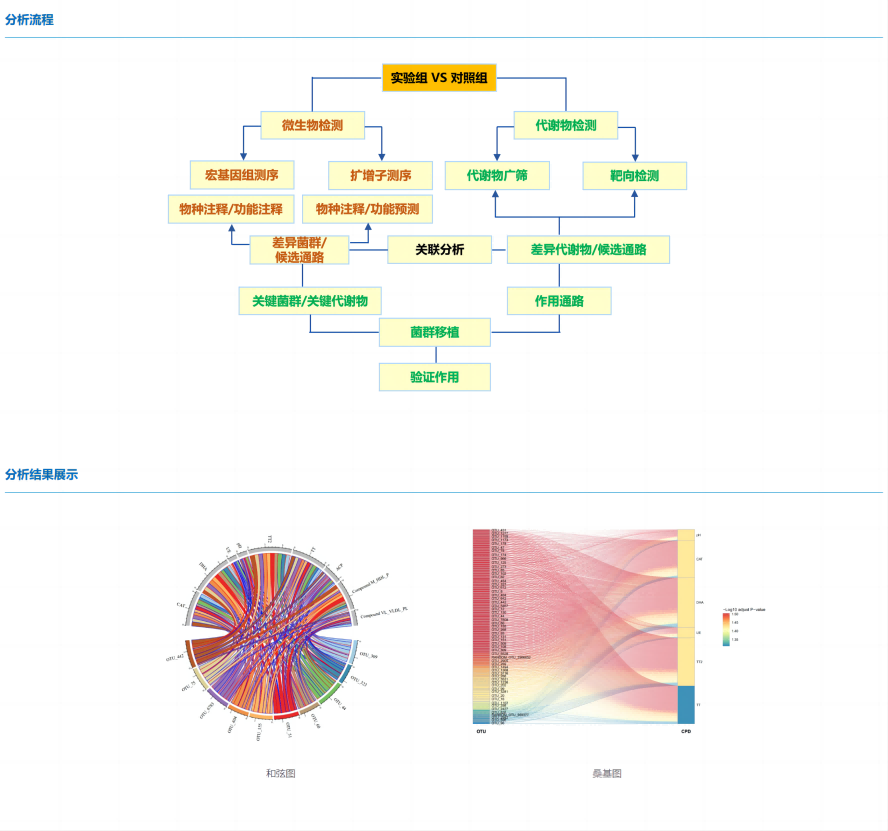
Microbiome, represented by microbial diversity and metagenomics, is used to study microbial community composition, diversity, and gene function information. Metabolomics, on the other hand, is the simultaneous qualitative and quantitative analysis of all low molecular weight metabolites of an organism or cell in a specific physiological period, and can study the relative relationship between metabolites and physiological and pathological changes.
It is difficult to comprehensively and systematically analyze the regulatory mechanism of complex physiological processes from a single omics data, and the joint analysis of multi-omics can jointly explore the potential regulatory mechanisms in organisms from different perspectives, and correlate the two omics data through various methods such as correlation coefficient, O2PLS and network diagram, which can jointly solve scientific problems from the level of species, genes and metabolites, and has a good application prospect in host physiology, disease pathology, drug pharmacology, etc.
Joint analytics benefits
l Data analysis from multiple perspectives;
l The results are more comprehensive;
l The conclusions of the study are more reliable;
l New thinking of correlation analysis.
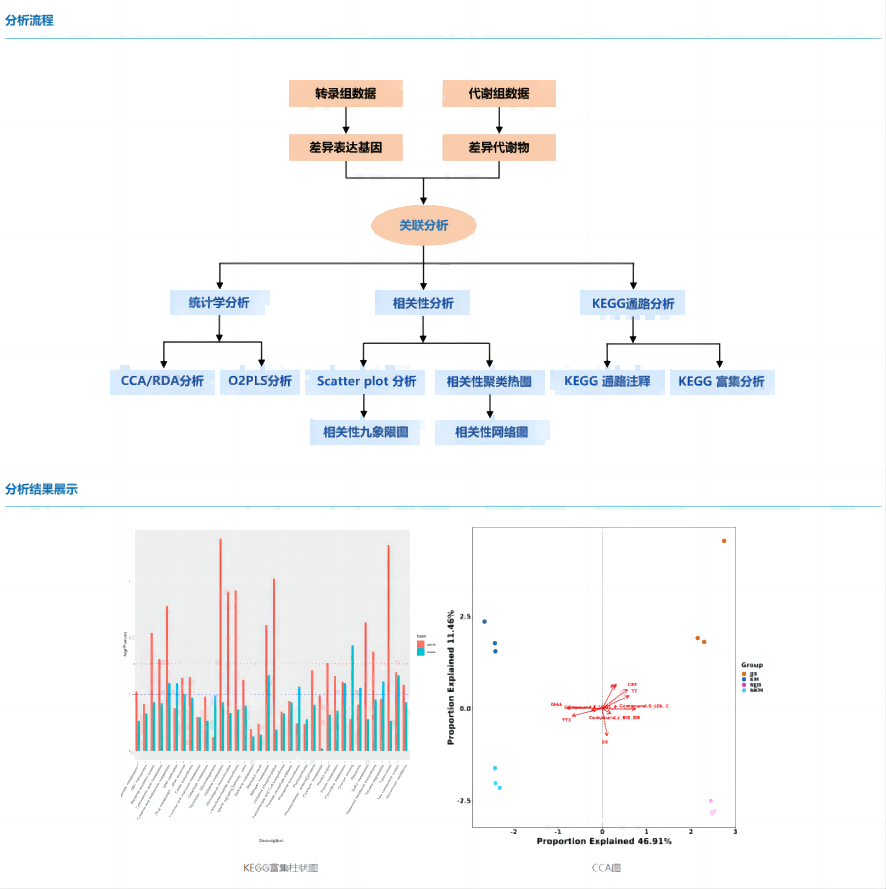
Transcriptome is a powerful tool for the study of functional genes, and a large number of differential genes and regulatory networks can be obtained by transcriptome sequencing, but it is difficult to determine the key pathways. The metabolome is the direct embodiment of phenotype, and the use of metabolomics data can reflect phenotypic changes, but it cannot explain the genetic mechanism that affects phenotype. Using transcriptome + metabolome multi-omics analysis, organisms can be studied at different levels, and the data of different omics also play a role in mutual validation, so as to comprehensively and systematically resolve complex biological processes.
The transcriptome can be analyzed by standard analysis procedures to obtain the analysis results of differentially expressed genes, and the results of differential metabolites and metabolic pathways can be obtained by metabolomics. By conducting joint analysis of differential genes and differential metabolites, calculating the correlation between differential genes and differential metabolites, and constructing a correlation network, the key genes that cause changes in metabolites can be identified and the key regulatory pathways can be determined. The conventional joint analysis mainly includes correlation analysis, KEGG pathway analysis, KEGG enrichment analysis, etc.
Joint analytics benefits
l Through the analysis of expression levels at different levels, it can make up for the lack of single omics data and noise interference;
l At the same time, biological problems can be explored from the two directions of "cause" and "effect", and the mutual verification effect is more obvious, so as to avoid false positives;
l Explain the correlation mechanism between molecular regulation and phenotype, and systematically and comprehensively analyze the function and regulatory mechanism of biomolecules;
l Remove the false from the massive data and keep the true, and screen out the key metabolic pathways, genes and metabolites for follow-up in-depth experimental analysis and application.

|
24-hour service hotline:028-87712800 |

|
service email:sctchj@188.com |

|
company address:No. 2507, Building 1, No. 8, Yuren West Road, Jinniu District, Chengdu, Sichuan Province |